Blog
How to Predict a Protein Structure with AlphaFold (ColabFold, No Install): A Beginner’s Guide
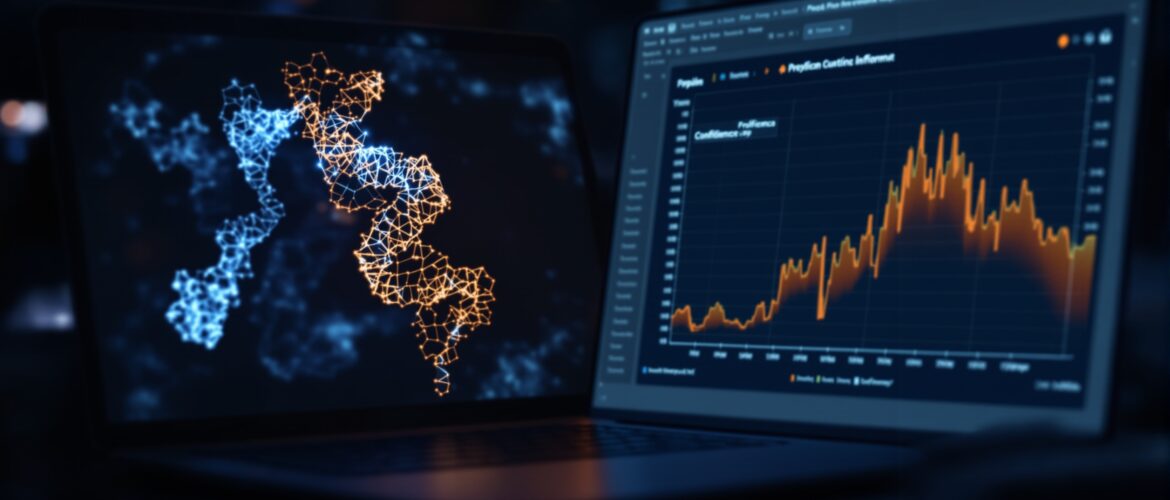
To predict a protein structure with AlphaFold, paste your amino acid sequence into ColabFold, a free Google Colab notebook that runs AlphaFold2 on a cloud GPU with no install. Run all cells, then read the pLDDT and PAE plots to judge whether the model is accurate enough to dock or simulate.
You have a protein sequence and no experimental structure, and the next step in your project needs a 3D model to dock a ligand or run a simulation. This guide walks an MSc student through folding that sequence with AlphaFold using ColabFold, then reading the confidence plots so you know whether the result is trustworthy. It fits into the wider computational biology skills roadmap, where structure prediction is usually the gate before docking and molecular dynamics.
What does it mean to predict a protein structure with AlphaFold?
Predicting a protein structure means going from the one-dimensional amino acid sequence to the three-dimensional coordinates of every atom, without ever crystallising the protein in a lab. AlphaFold is a deep learning system from DeepMind that does this by learning patterns from the tens of thousands of known structures in the Protein Data Bank and from evolutionary information in related sequences.
The reason this matters is that experimental structure determination by X-ray crystallography or cryo-EM is slow, expensive, and does not work for every protein. For most sequences you will meet in a project, no one has ever solved the structure. AlphaFold changed how accessible that gap is: at the CASP14 blind assessment in 2020 it reached a median accuracy score (GDT) of 92.4 across all targets, a level competitive with experiment for many proteins, as reported by Jumper and colleagues (2021) in Nature. That single result is why a predicted model is now a reasonable starting point for downstream work, provided you check its confidence first.
What is ColabFold, and how is it different from AlphaFold?
AlphaFold is the prediction method. ColabFold is a free, ready-to-run wrapper around it that lets you fold a sequence in a browser with no local install and no database downloads. It was built by Mirdita, Steinegger, and colleagues, who replaced AlphaFold’s slow sequence search with the much faster MMseqs2 search. In their paper, published in Nature Methods (2022), they describe it plainly:
“ColabFold offers accelerated prediction of protein structures and complexes by combining the fast homology search of MMseqs2 with AlphaFold2 or RoseTTAFold.”
That speed-up is real and large. The same work reports a search 40 to 60 times faster than the standard AlphaFold pipeline, which makes folding a single protein on a free cloud GPU take minutes rather than hours. For a student without a workstation or admin rights, ColabFold is the practical way in.
Before you run anything, it is worth checking whether the work is already done. Several routes exist to get a 3D structure for your sequence, and folding it yourself is only one of them.
| Method | What you need | Typical time | Best when |
|---|---|---|---|
| Experimental structure (RCSB PDB) | A solved structure of your protein or a close match | Minutes to find | An experimental structure already exists |
| AlphaFold DB lookup | A UniProt accession with a precomputed model | Minutes | Your exact protein is already in the database |
| ColabFold (this guide) | Just the amino acid sequence | About 15 to 60 minutes on a free GPU | No experimental or database model exists |
| Local AlphaFold install | Sequence, large databases, and a strong GPU | Hours to set up | High volume, offline, or private sequences |
| Homology modeling (SWISS-MODEL) | A homologous template structure | Minutes | A good template exists and you want a quick model |
Always check the AlphaFold Protein Structure Database first by searching your protein’s UniProt entry. If a model is already there, download it and skip the run. If it is not, or you have a mutant, a designed sequence, or a construct that differs from the deposited one, fold it yourself with ColabFold.
How do you predict a protein structure with ColabFold, step by step?
The whole run happens inside one notebook in your browser. You do not download AlphaFold, and you do not manage any databases. Below is the sequence of actions for a single protein chain (a monomer).
Step 1: Get your sequence in FASTA form
Have the amino acid sequence ready as plain one-letter code, for example the string starting MKT.... If you only have a UniProt or NCBI accession, open that entry and copy the sequence. Remove any header line, spaces, and numbers so you are left with just the letters. ColabFold needs the sequence itself, not a file.
Step 2: Open the ColabFold notebook
Go to the official ColabFold repository at github.com/sokrypton/ColabFold and open the AlphaFold2.ipynb notebook (the “AlphaFold2 using MMseqs2” link). It opens in Google Colab. Using the official notebook matters, because copies floating around the web can be outdated or altered.
Step 3: Confirm you have a GPU
In Colab, open Runtime > Change runtime type and make sure the hardware accelerator is set to a GPU (T4 is the common free option). AlphaFold on CPU alone is impractically slow, so this check saves you a wasted run.
Step 4: Paste the sequence and name the job
In the first form cell, paste your sequence into the query_sequence field and type a short jobname. Leave the default options for a first pass: the MSA mode using MMseqs2 against the UniRef and environmental databases, no templates, and num_relax set to 0. These defaults produce a standard model without extra steps.
Step 5: Run all cells and wait
Choose Runtime > Run all. ColabFold first builds a multiple sequence alignment by searching related sequences, then AlphaFold2 predicts five models, and finally the notebook draws the confidence plots. For a small protein this takes minutes; a few hundred residues can take longer. Do not close the tab, and keep the browser active so the free session does not time out.
Step 6: Download the results
When it finishes, the notebook produces a results zip file it offers to download. Inside are the predicted structures as PDB files, ranked by confidence, with rank_001 as the top model. You also get the coverage plot, the per-residue pLDDT plot, and the PAE plot. The rank_001 PDB is the file you carry forward, but only after you read those plots.
Want the guided, hands-on version?
Our live Molecular Modeling & MD Simulations cohort bootcamp takes you from zero to running real docking and MD workflows, with a portfolio project for your grad-school applications.
How do you read the pLDDT and PAE plots?
A predicted structure without its confidence scores is dangerous, because AlphaFold always returns a model even when it is guessing. Two outputs tell you how much to trust it: pLDDT and PAE.
pLDDT is a per-residue confidence score. As the AlphaFold Protein Structure Database states, “AlphaFold produces a per-residue confidence score (pLDDT) between 0 and 100.” High values mean the local structure of that residue is reliable; low values mean it may be wrong or intrinsically disordered. The colouring you see on the model (blue for high, orange and red for low) is this score mapped onto the chain.
| pLDDT score | Confidence band | What it means | Use for docking or MD? |
|---|---|---|---|
| Above 90 | Very high | Backbone and side chains reliable | Yes |
| 70 to 90 | Confident | Backbone modeled well, most side chains reliable | Usually yes, but inspect the binding site |
| 50 to 70 | Low | Treat with caution, often flexible regions | Only after careful checks or refinement |
| Below 50 | Very low | Frequently disordered or unstructured in isolation | No, do not treat as a fixed structure |
PAE is the Predicted Aligned Error. It is a two-dimensional heatmap that estimates, for every pair of residues, how confident the model is in their relative position and orientation. Dark, low-error regions mean the model places those parts confidently with respect to each other. PAE is what tells you whether two domains are reliably arranged, even when each domain on its own has high pLDDT. For a single compact domain you mostly rely on pLDDT; for a multi-domain protein you read the PAE to see if the domain arrangement is trustworthy.
Is the model good enough to dock or run molecular dynamics?
Judge the region you actually care about, not the whole-protein average. If you plan to dock a ligand, look at the pLDDT of the residues lining the binding pocket. If those are above 70, and ideally above 90, the local geometry is a reasonable basis for docking. A high average pLDDT with a floppy, low-confidence loop somewhere else does not disqualify the model for a binding-site study.
Two habits keep you honest. First, trim or flag long low-confidence tails and loops (pLDDT below 50) before docking, because a disordered terminus modeled as a fixed coil can block a pocket that is open in reality. Second, if you are heading into molecular dynamics, an energy-minimisation and equilibration step will relax small clashes, but simulation cannot rescue a wrong fold. When your pocket residues sit in the low band, consider whether an experimental structure or a homology model on a solved template is the safer input.
Once the model passes these checks, it becomes the starting coordinate file for the rest of the pipeline. From here you would prepare the receptor and ligand for docking, following our guide on preparing a protein and ligand for docking, then run the docking itself as covered in the molecular docking pillar guide. If your goal is dynamics instead, the model feeds straight into a GROMACS molecular dynamics workflow.
What are common ColabFold errors, and how do you fix them?
Most problems on the free tier come from resource limits, not from your sequence being wrong.
- “You are not connected to a GPU” or a very slow run: the runtime is on CPU. Open
Runtime > Change runtime type, select a GPU, then re-run all cells. - The session disconnects or hits a usage limit: free Colab caps GPU time and disconnects idle tabs. Keep the tab active, and if you are blocked, wait and try again later or split a long job into smaller runs. Very large proteins may simply exceed the free tier.
- Out-of-memory on a long sequence: free GPUs cannot fold very large chains. If your protein has clear domains, fold each domain separately, then study them individually.
- A shallow alignment with low coverage: the coverage plot looks sparse and confidence is low. This is not a bug. It means few related sequences exist for your protein (an orphan or a designed sequence), and AlphaFold has little evolutionary signal to work from. Read the result with extra caution.
When in doubt about a specific field or option in the notebook, follow the official documentation in the ColabFold repository rather than a third-party tutorial, because the notebook options change between releases.
Frequently asked questions
Is ColabFold the same as AlphaFold?
No. AlphaFold is the underlying prediction method from DeepMind. ColabFold is a free, browser-based wrapper that runs AlphaFold2 for you, using a faster MMseqs2 sequence search so you can fold a protein without installing anything. The structure you get is an AlphaFold prediction; ColabFold just makes it easy to obtain.
Do I need a paid Google Colab account or my own GPU?
No. The free Colab tier includes a GPU that folds most single proteins in minutes. Paid tiers give longer sessions and more powerful GPUs, which help for large proteins or many runs, but they are not required to follow this guide.
What pLDDT is good enough to dock or run MD?
Judge the residues you care about, not the average. For the binding pocket, aim for pLDDT above 70, and ideally above 90. Regions below 50 are often disordered and should not be treated as fixed structure. Always trim or flag low-confidence loops before docking.
Can ColabFold predict protein complexes and multimers?
Yes. The AlphaFold2 notebook supports multimers: you enter several sequences separated by a colon. For complexes the PAE plot becomes especially important, because it shows whether the predicted arrangement of the chains is confident, which pLDDT alone cannot tell you.
Is it acceptable to use an AlphaFold model for docking in a publication?
It is common and accepted, provided you report it honestly. State that the structure is an AlphaFold or ColabFold prediction, give the pLDDT of the binding-site residues, and describe any trimming or refinement. Reviewers expect the confidence of the region you docked into to be reported, not just an overall score.
Structure prediction is one skill in a longer chain that runs from a sequence to a validated model to docking and simulation. If you want to build that whole chain in order, the computational biology skills roadmap lays out the sequence and the tools for each stage.
Want the guided, hands-on version?
Our live Molecular Modeling & MD Simulations cohort bootcamp takes you from zero to running real docking and MD workflows, with a portfolio project for your grad-school applications.
Written by the StemSkills Lab team, with more than 10 years of combined experience in sequence and structural bioinformatics, drug discovery and design, and multiscale molecular modeling.