Molecular Modeling
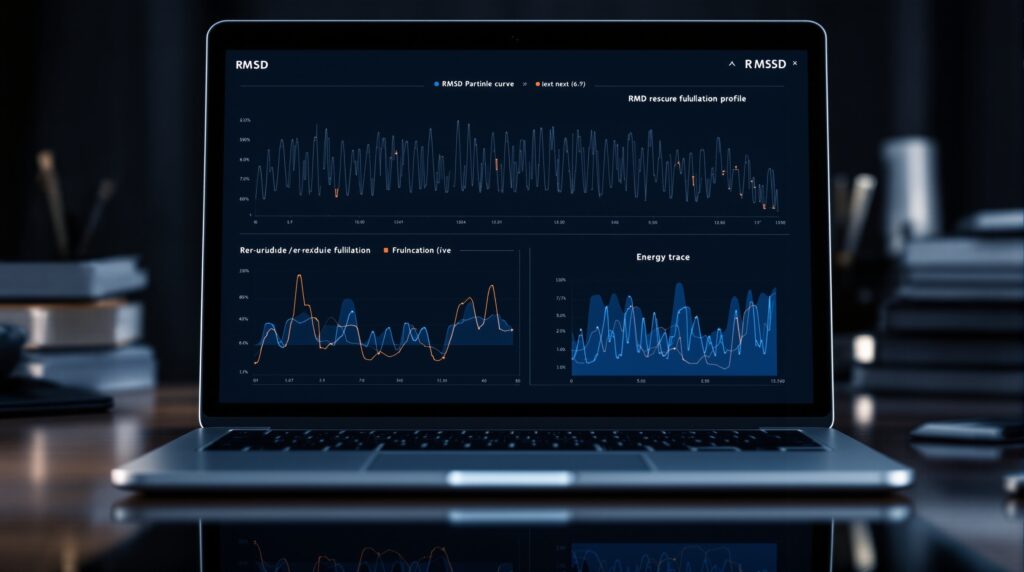
Parse GROMACS .xvg files in Python and plot RMSD, RMSF and energy with a reusable read_xvg function. Start making publication-ready figures today.

Turn a GROMACS trajectory into a 2D free energy landscape with gmx sham and PCA. Start mapping your protein’s conformational basins in kJ/mol today.

Learn to convert PDB to PDBQT, MOL2, SDF and SMILES with Open Babel and get your docking files ready today.

Check what hardware molecular docking and MD really need: laptop specs, why GROMACS wants a CUDA GPU, and the free Colab and HPC routes students can use.

Compare molecular docking and molecular dynamics: what a pose proves, what a trajectory adds, and why a defensible thesis project runs both in order.

Pick a defensible protein target and ligand set before you open AutoDock Vina. Follow the step-by-step selection checklist that survives a viva.

Pick a defensible MD simulation length and prove equilibration from RMSD, energy, temperature and pressure plateaus. Read the decision guide.

Turn your GROMACS run into a methods section a reviewer cannot reject. The exact parameters to report, real citations, and a worked example to adapt.
Turn a docked pose into a figure your supervisor accepts. Real PyMOL commands for cartoon-plus-sticks, labelled H-bonds, and a 300 dpi ray-traced render.

Stop copy-pasting em.mdp and md.mdp. Learn which GROMACS .mdp parameters you must choose deliberately, which to leave at defaults, and what breaks when you get them wrong.